A basic microbiome analysis¶
QIIME¶
Note
| Required OS: | OS x or Linux |
|---|---|
| Software: | Qiime 1.9 |
| Documentation: | Qiime tutorial |
| Author: | This document was created by Saranga Wijeratne |
File formats¶
This section includes descrption of varies file formats, including Qiime scripts, and parameters files. Read more here
Qiime Script index: Index of all the scripts used in Qiime.
Metadata mapping files: Metadata mapping files provide per-sample metadata.
Tip
A metadata mapping file example is given here. Read the section carefully. If you are planning to create the mapping file by hand, read this section.
Biom file: OTU observation file. Read more here.
Download the files¶
For this tutorial, we will use Mothur tutorial data from Schloss Wiki. These data are 16s rRRNA Amplicons sequenced with Illumina MiSeq.
1. Create folders
Make a new directory MCICQiime and then cd to move into the dirctory.
$ mkdir MCICQiime $ cd MCICQiime
2. Download data
Download data from Schloss Wiki
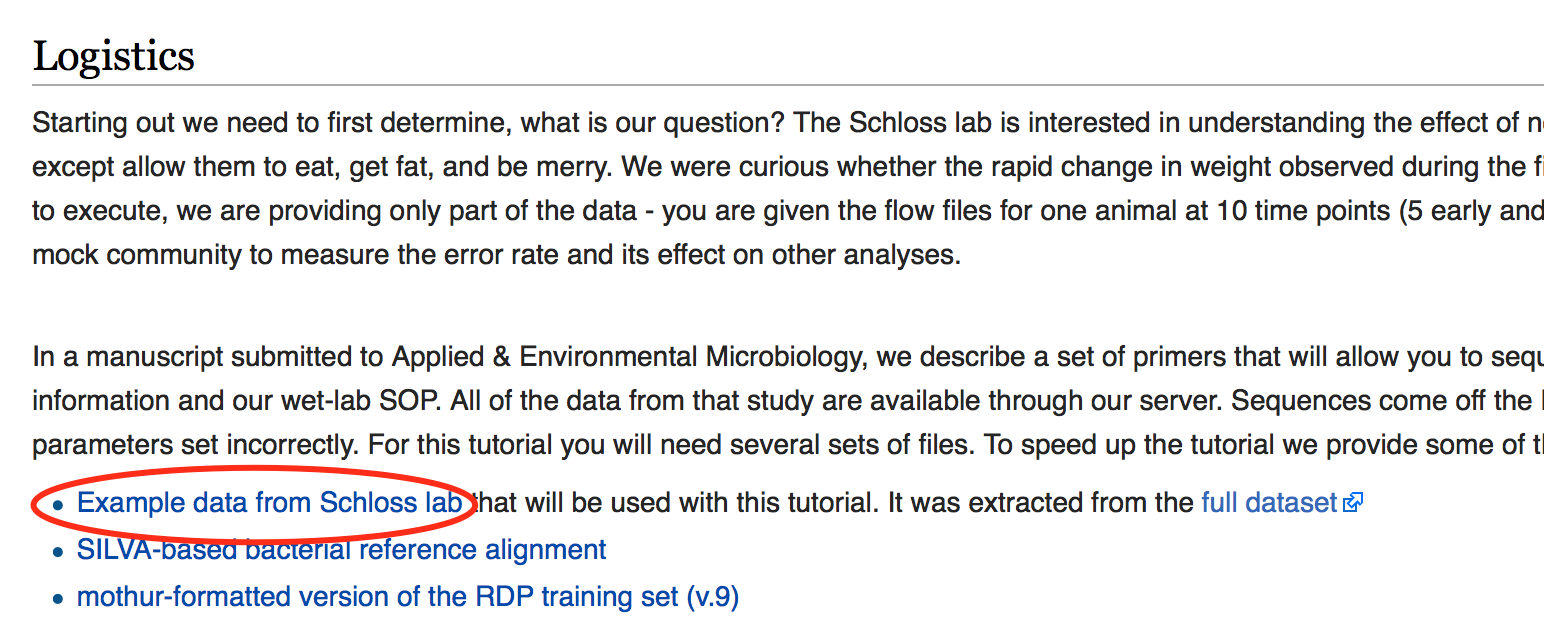
For this tutorial download only dataset shown in the image below (i.e Example data from Scholoss lab).
Inside the MCICQiime dir, issue the following command to get the data.
The data has been zipped, and we will use unzip -j to extract all the files to same directory we are in right now.
Rename the files for downstream analyses:
And explanation of the preceding commands:
for f in *.fastq;reads any file that ends with .fastq, one at a time.dostarts the body of the for loop.mv $f do mv $f ${f%%_L*}.fastq;rename $f (i.e F3D0_S188_L001_R1_001.fastq) to ${f%%_L*}.fastq (i.e F3D0_S188.fastq)donefinishes the loop.
3. An explanation of the data
The files and experiment are described in the Schloss Wiki.
Because of the large size of the original dataset (3.9 GB) we are giving you 21 of the 362 pairs of fastq files. For example, you will see two files:F3D0_S188_L001_R1_001.fastqandF3D0_S188_L001_R2_001.fastq. These two files correspond to Female 3 on Day 0 (i.e. the day of weaning). The first and all those with R1 correspond to read 1 while the second and all those with R2 correspond to the second or reverse read. These sequences are 250 bp and overlap in the V4 region of the 16S rRNA gene; this region is about 253 bp long. So looking at the files in theMiSeq_SOPfolder that you’ve downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mock community. You will also seeHMP_MOCK.v35.fastawhich contains the sequences used in the mock community that we sequenced in fasta format.
GBSv2 pipeline plugins¶
| Plugin | Description |
|---|---|
| GBSSeqToTagDBPlugin | Executed to pull distinct tags from the database and export them in the fastq format. More |
| TagExportToFastqPlugin | Retrieves distinct tags stored in the database and reformats them to a FASTQ file. More |
| SAMToGBSdbPlugin | Used to identify SNPs from aligned tags using the GBS DB. More |
| DiscoverySNPCallerPluginV2 | Takes a GBSv2 database file as input and identifies SNPs from the aligned tags. More |
| SNPQualityProfilerPlugin | Scores all discovered SNPs for various coverage depth and genotypic statistics for a given set of taxa. More |
| UpdateSNPPositionQualityPlugin | Reads a quality score file to obtain quality score data for positions stored in the snpposition table. More |
| SNPCutPosTagVerificationPlugin | Allows a user to specify a Cut or SNP position for which they would like data printed. More |
| GetTagSequenceFromDBPlugin | Takes an existing GBSv2 SQLite database file as input and returns a tab-delimited file containing a list of Tag Sequences stored in the specified database file. More |
| ProductionSNPCallerPluginV2 | Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF or HDF5 format. More |
GBSv2 pipeline¶
1. Load the Tassel 5.0 module
1 | $ module load Tassel/5.0
|
2. Useful commands
To check all the available plugins, type:
1 | $ run_pipeline.pl -Xmx200g -ListPlugins
|
To check all the parameters for a given plugin, e.g. GBSSeqToTagDBPlugin, type:
1 | $ run_pipeline.pl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
|
Tip
Users are recommended to read more about GBS command line options here. Page 1-2
3. File preparation
Create necessary folders and copy your raw data (fastqs), reference file and key file to appropriate folder:
1 | $ mkdir fastq ref key db tagsForAlign hd5
|
4. Commands for the pipeline
1 2 3 4 5 6 7 8 9 | $ run_pipeline.pl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k key/Tomato_key.txt -e ApeKI -db db/Tomato.db -kmerLength 85 -mnQS 20 -endPlugin -runfork1
$ run_pipeline.pl -fork1 -TagExportToFastqPlugin -db db/Tomato.db -o tagsForAlign/tagsForAlign.fa.gz -c 5 -endPlugin -runfork1
$ cd ref
$ bwa index -a is S_lycopersicum_chromosomes.2.50.fa
$ cd ../
$ bwa samse ref/S_lycopersicum_chromosomes.2.50.fa tagsForAlign/tagsForAlign.sai tagsForAlign/tagsForAlign.fa.gz > tagsForAlign/tagsForAlign.sam
$ run_pipeline.pl -fork1 -SAMToGBSdbPlugin -i tagsForAlign/tagsForAlign.sam -db db/Tomato.db -aProp 0.0 -aLen 0 -endPlugin -runfork1
$ run_pipeline.pl -fork1 -DiscoverySNPCallerPluginV2 -db db/Tomato.db -sC "chr00" -eC "chr12" -mnLCov 0.1 -mnMAF 0.01 -endPlugin -runfork1
$ run_pipeline.pl -fork1 -ProductionSNPCallerPluginV2 -db db/Tomato.db -e ApeKI -i fastq -k key/Tomato_key2.txt -kmerLength 85 -mnQS 20 -o hd5/HapMap_tomato.h5 -endPlugin -runfork1
|